Jean-Martin Charcot, a French Neurologist, (1825-1893) widely recognized as the Father of Neurology, reported a series of 20 patients whose illnesses were characterized by dramatic and progressive loss of muscle strength with associated muscle atrophy in 1874. Charcot personally developed the term Amyotrophic Lateral Sclerosis based on the unique autopsy finding in the brain of several of his patients. (2) Initial recognition of a single patient with ALS type findings is attributed to Dr. Charles Bell in London, England, 1824, a half century earlier.
ALS most frequently is diagnosed in white males between ages 40 and 70. ALS is rarely diagnosed in patients less than 20 years old. Diagnosis to death is less than 60 months in 90% of patients with ALS.
Alois Alzheimer, (1864-1915) a German Psychiatrist and Neuropathologist, described the clinical course of his unique patient: Auguste Dyer (1850-1906) in 1907 in Bavaria, Germany. Her clinical findings were characterized by dramatic loss of her memory associated with increasingly severe psychotic and erratic behavior. Her autopsied brain, personally studied by Dr. Alzheimer, a neuropathologist, himself many years later demonstrated unique cerebral cellular changes and nerve tangles now diagnostic of its namesake disease.
Her husband had institutionalized his wife in the local insane asylum because of her increasingly erratic and psychotic behavior rather than because of her memory loss. It was here that Dr. Alzheimer met Auguste Dyer for the first time several years earlier.
Dr. Alzheimer was so intrigued by the rapid deterioration of Auguste’s D’s memory and her progressively erratic behavior, that he personally recorded, in his own handwriting, a series of simple questions and her totally inappropriate, ever changing answers. One of Dr. Alzheimer’s colleagues referred to Auguste Dyer’s illness as “Alzheimer’s disease” shortly after he presented her clinical course at a local medical meeting in Bavaria, Germany in 1906-7. Best estimates are that her memory loss and erratic behavior began more than 5-10 years prior to her being institutionalized at age 50.
It appears purely ironic that the name of a German Psychiatrist would now be associated with a disease feared by every educated senior. The incidence of Alzheimer’s is projected to increase commensurate with the ever increasing aging population of senior citizens. There is so little hope for preventing the development of Alzheimer’s or discovering an effective treatment that at least one financial planning institution has already projected the incidence and cost of this disease 30-40 years into the future.
Today Auguste Dyer would be classified as having “early onset Alzheimer’s“ as her disease became manifest prior to age 50. The significance of this diagnosis, (early onset Alzheimer’s) a century later, is still not entirely clear. August Dyer is cited as having a daughter herself who apparently had a child of her own. Both were considered to be normal when last seen. Follow up records of Auguste’s granddaughter are unavailable.
Alzheimer’s disease is now considered to be the fifth-sixth leading cause of death in America, and certainly among the most expensive diseases to treat, manage and care for, currently known to exist.
With respect to ALS, it is easy to calculate that when viewed as a distinct neurodegenerative brain disease, it will soon be nearly 200 years that we have been aware of its existence and 112 years that we have been aware of Alzheimer’s in addition to a variety of other forms of degenerative brain diseases.
Cumulatively these neurodegenerative diseases have been known to exist for three centuries. Nevertheless they are each still without known cause or cure. Why?
Most serious students of neurodegenerative diseases consider ALS and Alzheimer’s to be distinct and unrelated diseases. These diseases differ in several major respects: Alzheimer’s affects neurons associated with memory. ALS affects motor neurons and their peripheral nerves directing motion.
Sachatello chooses to consider both diseases to be “related”, if not essentially identical, primarily because of the similar pattern of glial-neuron cell death. All neurodegenerative brain diseases share several characteristic features: specific glial neuron complexes insidiously lose function, degenerate and die. Similarly, these neurodegenerative brain diseases have a progressive course, leading to profound disability and a shortened life span for each afflicted patient.
This theory, as proposed, has no explanation for the selection of targeted receptor neurons in separate but specific areas of the brain, unless they are designated by the unique similarity of structural and spatial configuration of their specific hepatic generated neurotoxins.
In the complete and total absence of any other comprehensive and inclusive theories concerning neurodegenerative diseases, the author again proposes the following theory: (his best educated guess), in simple declaratory (but unproven) sentences. (1-3)
“The glial neuronal cell damage and clinical course seen in both Alzheimer’s and ALS result from the inability of an ageing, but histologically normal liver, to completely metabolize normal substrates it encounters daily, resulting in a variety of incompletely metabolized byproducts, some of which are neurotoxic.
These hepatic generated neurotoxins are the direct proximate cause of neurodegenerative brain diseases. The quantity and variety of neurotoxic metabolites produced, may well determine whether any given patient develops ALS, Alzheimer’s or other type of Primary Dementias”.
Amplification of this hypothesis will readily support the clinically observable following propositions for neurodegenerative brain diseases.
- “In general, the earlier the onset in life, the more severe the disease: the more rapid the progression, the shorter the functional lifespan of the brain or other vital motor nerve connections.” (This proposition is based on the premise that the higher concentration of neurotoxins, the more the extensive resulting nerve damage will develop. Likewise, the more extensive the resulting nerve cell damage, the more severe the resulting illness; and the more rapid the death of vital nerves and the patient himself.) Lou Gehrig’s brief illness, prior to his death supports this proposition for ALS quite accurately.
- The incidence of Alzheimer’s increases with age due to the fact that hepatic generated neurotoxins are being continuously produced, even in as yet undiagnosed patients. The older the patient, the higher the probability, that patients have some degree of memory loss. This proposition appears to be confirmed by observable, progressive memory loss in the majority of elderly patients.
- The rate of progression of memory loss, erratic or psychotic behavior, increases with age, due to the continued production of hepatic generated neurotoxins with resultant neuronal-glial cell death.
- Delayed recognition of Alzheimer’s, (the duration of the prodromal period) may be due to a number of factors:
- 4A. the total number of neurons at risk: (e.g.) those in the cerebral cortex)
- 4B. the quantity of neurotoxins produced
- 4C. the relative toxicity and type of neurotoxins produced
- 4D. prior innate intelligence and level of education
- 4E. dietary factors (as yet undetermined)
- 4F. prior cerebral damage (e.g.) individuals who have ingested specific hepatotoxins, (excess amounts of alcohol (e.g.) over a prolonged period of time) would appear to be much more likely to manifest memory loss much earlier than those with minimal prior ingestion of hepatotoxins.
In summary, it is much more likely that an individual with greater innate intelligence would be more likely to manifest Alzheimer’s type symptoms later in life than a person with much less innate intelligence. Similarly individuals who have damaged their liver by life style choices: “alcohol abuse” would most likely show signs of Alzheimer are much earlier in life.
The best explanation for the disparity in the development of these diseases would be to estimate the number of neurons at risk. Alzheimer’s would primarily affect memory neurons, the exact number of which is currently undetermined, but is estimated to be in the multiple billions, if not more. ALS would be limited to specific motor neurons. It would appear only logical that ALS would have a much shorter prodromal phase simply based on the greater ratio of neurotoxins to targeted motor neurons. Cumulative motor neuron destruction must reach a critical mass to produce diagnostic clinical findings in ALS. - The extended duration of the prodromal phase in Alzheimer’s is best explained by considering that the causative neurotoxins in Alzheimer’s are relatively weak individually. Relatively weak neurotoxins would require an additional extended period of time before they are sufficiently numerous to destroy a critical portion of the uncountable billion number of targeted memory glial-neurons complexes.
- This theory predicts that the duration of the prodromal phase of Alzheimer’s will prove to be substantially longer than that currently estimated as soon as this disease is diagnosable by simple blood tests. It is highly probable that there will be a time in the future when the length of the prodromal phase can be calculated with precision by objective laboratory studies. The author anticipates that diagnostic laboratory findings will be measurable long before the patient is an adult.
Admittedly, these assumptions are extraordinarily speculative and certainly will be heretical to most investigators. Nevertheless, they provide a testable hypothesis, never provided before, by any other previously published theory attempting to identify the inciting cause and progressive clinical course of Neurodegenerative Brain diseases. - If one documents the first manifestation of ALS, solely by the initial and measurable loss of muscular coordination, (the ability to hit a ball, traveling at 80-90 mph, with a stick), Gehrig’s prodromal course would appear to be in the range of a year or two at most.
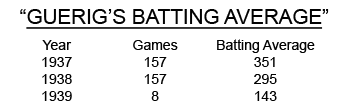
Estimated Onset of Gehrig’s ALS: (late summer 1938 - Early Spring, 1939)
Gehrig’s batting average in his first 79 games was slightly less than in his last 78 games in his 1938 season. The fact that Gehrig had only 4 singles in the 1938 World Series adds credence to his insidious loss of muscle strength in late summer 1938.
There is no mention of Gehrig having any type of neuromuscular incoordination or dysfunction during his 1938 season. He is reputed to have told one of his team mates he did not feel as strong as usual, in mid-season 1938.
A few ALS patients have an unexplainable variation in the rate of progression of their disease: a very small number of patients appear to live 2 to 5 times longer than others. Many serious students of ALS cite specific instances during which there is minimal progression of the underlying disease for months or even years, without explanation. Unfortunately there is minimal published data confirming that these specific patients actually had ALS.
This theory has a possible explanation for the variation in longevity. Hepatic generated neurotoxins could easily vary in both quantity and the strength of their relative neurotoxicity due to as yet undetermined dietary factors.
Primary Dementia: It is only natural that there would be a number of various manifestations of primary dementias. Some neurotoxins would primarily affect one aspect of behavior, while others would be more likely to affect other aspects of memory or various other forms of antisocial behavior. These differences would be dependent on a combination of the chemical structure and quantity of neurotoxins produced in addition to a patient’s innate intelligence, personality and prior education.
This theory further predicts that the chemical structure, spatial configuration and quantity of specific neurotoxins will prove to correlate directly with the characteristic type of observed dementia.
Initial Posted on line: May 26, 2015
Most recent revision: 8/01/2018
← Go Back to Previous Section Continue to Next Section →
A logical extension of the liver brain theory, should it prove to be correct, is a simple, but another extremely heretical idea! Proposing this theory will lead many professionals to openly question the sanity of the author.
4B-1 Based on the simple, but heretical, premise that the proximate cause of both ALS and Alzheimer’s are hepatic generated neurotoxins it is only logical to conclude that liver transplant in a patient afflicted with ALS will very likely prevent the inevitable progression of muscle weakness giving an ALS afflicted patient a more normal life expectancy.
There is minimal probability that any benefit would be seen in Alzheimer’s primarily because of the extended duration of the prodromal period prior to diagnosis of this disease. A very high percent of the memory neurons in Alzheimer’s patients would have already died during the prodromal period.
It should be a foregone conclusion that when a young intelligent otherwise healthy patient is diagnosed with ALS, and is informed he has a 95-100% probability of dying within two to four years; he might opt for an experimental liver transplant. In the past decade, the number of liver transplants (replacements) has remained relatively constant in the range of 5,000 – 6,500 per year. Average survival post liver transplant is in the range of 10-20 years or more greatly dependent on the patient’s willingness to take care of themselves.
Liver transplants are painful, expensive and fraught with unforeseeable complications. At present they are reserved exclusively for patients with terminal end stage liver disease who would be dead without one within days or weeks.
In view of the fact this proposal for an experimental liver transplant is so far outside what most might consider rational thought the author feels obligated to expand on the pros and cons to any prospective patient considering such a decision.
When an attending physician speaks to a patient and his family with ALS he is obligated to present all relevant facts: ALS is an inevitably fatal disease still without any known or effective treatment nearly 200 years after first being recognized in a single patient in 1824. In a few Scandinavian countries suicide is acceptable alternative treatment for ALS patients. (10)
The author conceived the theory that a liver transplant will arrest the progression of muscle weakness in ALS purely by deductive reasoning. There are no experimental animal models in which to test this theory in a laboratory.
Should the premise of the liver brain theory be validated as the author believes it will be, a new healthy liver will not continue to produce the hepatic generated neurotoxins he theorizes to be the proximate cause of ALS. A successful liver transplant in an ALS patient may grant ALS patients a more normal life expectancy.
4B-2. Approximately 6,000 to 7,500 liver transplants have been performed annually in the past decade in the US. The number of transplants is limited only by the number of healthily transplantable livers. Because of the increasing paucity of transplantable livers relative to their need, the recent trend is for a family member to donate part of his liver to a related patient near death from end stage liver disease. Current estimates are that 5-10 % of liver transplants are now from live donors. The first patient to receive part of her dad’s liver recently celebrated her 25th post-transplant anniversary. The human liver is the only organ that can regenerate itself.
Total debunking of this heretical theory will have major benefit: it will allow others to be willing to propose new ideas, without acquiring the stigma of being labeled as “crazy” as this author anticipates he will be if this theory is repudiated by several human experiments.
Although the incidence of Alzheimer’s is approximately 300 times more common more than ALS (5.7 million versus15- 20,000), Alzheimer’s patients have no easily quantifiable disease related factors amenable to measurement with assured reproducible accuracy. As such, patients with Alzheimer’s disease are not suitable for most initial clinical studies.
Patients with ALS have a distinct and unique disease related measurable factor, “progressively declining muscle strength.” Muscle strength can be measured repetitively with decimal like precision over time. Even more important, these measurements can be made with simple readily available noninvasive techniques at minimal cost.
Biting strength, merely another form of muscle strength should be easily quantifiable. It specifically measures the primary muscles of mastication: “temporalis, medial and lateral pterygoids.” This measurement is unique in that these muscles are innervated by an intracranial nerve: the third (V3) division of the trigeminal nerve. By recording these measurements and systematically plotting their natural course of muscle weakness over a period of time, one can measure the decline in muscle strength and function of both somatic and cranial nerves simultaneously.
As noted elsewhere, these simple measurements could have been completed for a pittance more than 50 years ago. Even today there is minimal, if any, published data recording whether cranial or somatic nerves degenerate at the same or differing rates in ALS patients.
The author suggest it would be far more advantageous and cost effective initially to study ALS in specific detail, rather than embark on a random, directionless and most likely futile study of Alzheimer’s.
The very first clinical studies in ALS patients must attempt to measure, quantify, retard or alter the rate of progression of loss of muscle strength both by age of onset and sex.
Several simple dietary studies in patients with ALS are mandatory.
(A) Does altering the amount of caloric intake? or
(B) Varying the quantity of specific proteins affect the rate of muscle loss and strength?
Simply confirming that the rate of progression of ALS can be altered should provide an additional impetus for further evaluation of dietary factors. If such simple studies confirm that diet alone, can alter the rate of progression, in matched controls in patients with ALS, this seemingly insignificant study will provide the first real evidence ever; ALS might be amenable to treatment.
This line of reasoning is addressed in detail in Section 12:
Another simple question pertinent to diets is: Do absolute vegetarians have the same, lesser or greater incidence of ALS in particular? An equally important question: If an absolute vegetarian does develop ALS, does it progress at the same or greater rate than in non-vegetarians?
It is imperative to appreciate that there may be substantial differences between various types of vegetarian diets as determined in a recent study of the incidence of colorectal cancers in vegetarians (12).
The liver brain theory predicts that certain types of vegetarians (yet to be determined) would have both a decreased incidence of ALS as well as a decreased rate of progression compared to non-vegetarians. The exact type of vegetarian diet that might influence the rate of progression of ALS can be determined only by additional study using the parameters noted in the references cited. (12)
Another simple inexpensive clinical study, specifically related to Alzheimer’s is as follows: does the amount, duration, frequency or age of onset of alcohol abuse, correlate with the age of onset, or incidence or rate of progression of Alzheimer’s? The liverbraintheory states the answer should be self evident: suggestive evidence should be manifest in almost every homeless shelter!
Current studies suggest that the overwhelming majority, if not nearly 100%) of intra cranial neurons are formed in utero.
Although there appears to be minimal agreement concerning the ratio of glial cells to cerebral neurons in various and specific areas of the brain, this author would favor the proposition that neurotoxins precipitate the death of glial cells that lead to Alzheimer’s by the simple process of deprivation of essential nutrients to specific glial cells anatomically adjacent nutritionally dependent neurons.
In Pursuit of the Proximate Cause of ALS
The simplest and least expensive initial laboratory experiments would entail obtaining liver tissue from recently diagnosed or deceased patients with all types of neurodegenerative brain diseases in addition to patients with alcoholic cirrhosis.
Aliquots of fractioned homogenized liver specimens should be added to glial-nerve cell cultures. The end point would be to document the presence of neuronal-glial degeneration, deformation or cell death in otherwise normal glial cells once previously thriving in cell culture. Unanticipated finding of such glial changes, particularly death, would add credence to this theory that glial cells are the primary target of hepatic generated neurotoxins.
The importance of obtaining specimens from younger patients with neurodegenerative brain diseases cannot be over emphasized. This theory predicts that the younger the patient, the greater the concentration of neurotoxins, or that those that are present, are much more neurotoxic.
May I ask each serious researcher in this field a simple question? Could you be opened minded enough to accept the possibility, however remote, that Alzheimer’s is primarily a liver disease, and that associated neurodegenerative changes are secondary to an aging liver producing neurotoxins?
The very same question applies to ALS. It appears to be a functionally related disease. Intense study of this motor neuron disease will open an entirely new field of research, one that is quantifiable with decimal like precision.
Additionally I humbly suggest, to each and every student of neurodegenerative brain diseases, if you are unwilling to consider this theory worthy of further study, you have a personal obligation to develop one of your own! We would be honored to publish your theory here without further review.
An initial two pronged effort is mandatory, especially in young recently diagnosed patients with ALS. While one group seeks to verify the presence of neurotoxins, as outlined above, another team should attempt to remove them by vigorous and repetitive plasmapheresis using standard techniques.
It is more than reasonable to speculate that a certain percent of neurotoxins would be circulating continuously. Some neurotoxins, already intracellular, but unattached, might possibly be eluted due to a difference in concentration gradients, resulting from repetitive plasmapheresis. Any statistically significant data, confirming a decrease in of the rate of progression of ALS must be pursued with renewed vigor.
In summary, a few simple inexpensive studies may well provide the first real clues to the cause of either or both of these neurodegenerative brain diseases, now recognized to have existed in the prior two centuries. Should only a tiny fraction of this hypothesis be confirmed, both diseases could possibly be of historical interest only in less than a decade or less.
Is it conceivable that vigorous “ mental gymnastics ‘”can delay, retard or possibly preclude the development of Alzheimer’s?
Could it be reasonable to ask a simple question? Can maximum cognitive function (E .G) playing chess, backgammon or bridge) stimulate the replication of glial cells rendering their anatomically associated neurons more efficient in processing increased data while providing them an additional level of sustenance.
Many students of Alzheimer’s appear to have adopted their own plans to reduce their personal likelihood of developing this terrible disease: vigorous mental gymnastics on a regular basis: reading, writing, playing chess, or other demanding cognitive functions such as serious investing of one’s own money.
No better example of this latter endeavor could be found in that of the recent public virtuoso performance on TV station CNBC by one the worlds’ most sophisticated investors and wealthiest men: Warren Buffet. This 85 year old gentleman recently presented his overview of the world’s economy and in doing so discussed the pro and cons of investing in a variety of public companies, while citing specific balance sheet figures, essentially hieroglyphics to all but a few. In addition he proudly admitted to playing chess (online) at least 10 hours a week.
Is Buffet’s exceedingly astute mental function at age 85, purely happenstance, a derivative of family genetics, (His dad was a Congressman.) or is it demonstrable and definitive proof that there is real benefit in routine mental gymnastics?
The exact same question can be asked of his 95 year old business partner, Mr. Charles Munger. This gentleman is cited as being one of Mr. Buffet’s closest advisors. His mental gymnastics appear to have kept his thought processes equally astute.
From a strictly personal point of view, these assumptions appear to have real merit. Unfortunately, the author was unable to find a single controlled study addressing or supporting this premise.
Here again it is very sad to recognize that a near definitive answer to this simple question could have been determined for only a pittance as long as a half century ago. How? Simply by comparing the statistical probability of developing Alzheimer’s in two age controlled groups: those who read, write and think versus those who snooze, snore and sleep!
The very concept that merely using one’s brain could possibly influence the probability of developing a neurodegenerative brain disease appears biologically improbable. Nevertheless it is imperative to recognize that the cellular composition of the brain is substantially different than any other organ system. The brain basically has two distinct types of cells, neurons and glial cells. There is little evidence that neurons retain their ability to replicate postpartum. Glial cells definitely do retain this ability on a near life time basis. Is it reasonable to assume that vigorous mental gymnastics have the ability to stimulate the replication of glial cells and in doing so preserve and prolong the life and function of their anatomically adjacent nutrient dependent neurons?
I openly hypothesize: (my best educated guess) is that the inciting cause of neurodegenerative diseases originates in the patient’s own liver, leading to a cascade type reaction possibly playing a major role in formation of tau proteins or other as yet to be identified neurotoxins.
The very first question any neurologist-- physician might ask: Why would a retired octogenarian surgeon have wasted his time for the previous 8 years plus years seeking a solution to these complex diseases that have defied a definitive answer for the past two centuries?
The first answer is simple: I had no other choice, for reasons sufficient to myself, but to proceed to study Alzheimer’s disease in detail. I did so, but from an entirely different perspective from that of a budding neurology resident hoping to pass his boards the first time around. I studied these diseases from the perspective an experienced, retired knowledgeable surgeon:
- Everything has a cause!
- What is the cause (etiology) of Alzheimer’s or ALS?
- Why has it been so difficult to generate even a simple clue, relative to the etiology of neurodegenerative brain diseases for the past two centuries?
In the author’s opinion, it is just a question of a relatively short time, before someone with an open mind, using the scientific method, will soon discover the answers to questions 2 & 3. The more I read, the more I was convinced of two things in particular:
- The cumulative knowledge of the causation of both ALS and Alzheimer’s is minimal at best.
- Shared ignorance is the rule among the so called “experts“.
Another reasonable conclusion appears to be quite apparent: it is highly unlikely that any meaningful progress can be made in the study of Alzheimer’s disease simply because of the fact there is nothing to measure with any degree of certainty on a repetitive basis.
Variables such as innate intelligence, education, family support, including financial resources, and the ready availability of medical care are so significant that it is highly unlikely any reliable control groups can be established.
The importance of innate intelligence cannot be over emphasized. Were one individual tested be in the upper decile of innate intelligence be compared with a similarly aged individual known to be in the lowest decile of intelligence, sustain the loss of 10% of memory neurons could very well result in a dramatically different outcome. The more intelligent individual would most likely be recognized as being “less sharp” than usual. The less intelligent individual could possibly require institutionalization!
Patients classified with Primary Dementias are much too amorphous as a group to warrant further intensive study at this time. To do so would be folly and a waste of valuable resources.
A logical conclusion of this theory would predict that is very likely that the causal factors of ALS will be recognized long before the etiology of Alzheimer’s is even imagined. Restating this proposition in the NEGATIVE, in the author’s opinion: The proximate causes of Alzheimer’s will never be determined prior to elucidating the proximate cause of ALS.
Muscle Strength can be measured repetitively, with decimal like precision, in response to a variety of stimuli and medical interventions. There are no similar disease related manifestations of Alzheimer’s lending themselves to similar measurements.
In the author’s opinion the definitive explanation for the proximate cause of Alzheimer’s will include the following sentence: “The proximate cause of Alzheimer’s disease is directly attributable to: primary, age related, hepatocyte dysfunction”.
Charles R. Sachatello MD FACS
Posted on line: 3/20/2015 and revised multiple times thereafter
2/14/2016--- 08/03/2018
← Go Back to Previous Section
Continue to Next Section →